
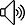
1.下載基因組注釋文件,選擇對應的版本:?ftp://ftp.ncbi.nlm.nih.gov/genomes/Homo_sapiens/ARCHIVE/BUILD.37.3/GFF/?
2.GTF 為General Transfer Format ,熟悉格式?http://www.huoyunjn.com/wuliuxinwen/2/33709819.htm。
第三列 feature ?- 后面start和end之間區域代表的特征,如果此區域是基因,則此處為gene,如果是外顯子,則為exon,如果是轉錄本,則為transcript,如果是非編碼RNA則為lncRNA,如果是重復序列,則為TE,等等,主要表明這一塊區域的特征。
3.每一個transcript對應的exon,所有長度加起來就是這個轉錄本的長度。與這個transcript后面的兩列相減是有差別的。
4.用python 字典來統計每個轉錄本的長度。
import pandas as pd
import pdb
df = pd.read_table(r'C:\Users\guosheng\Desktop\out.gff',sep = '\t',header= None)
out=open('./out.txt','a')
df =df[df.iloc[:,2].str.contains('exon')] #提取第三列為exon的行
df['diff'] =df.iloc[:,4]-df.iloc[:,3]+1 #每個外顯子的長度
name = list(df.iloc[:,2]) #把data.frame中的一列轉換為list
des =list(df.iloc[:,8])
length = list(df['diff'])
dic ={}
for index,value in enumerate(name):
key=des[index].split(';')[-1].split('=')[-1] #獲取每個轉錄本的名字
old=0
new=length[index]
if dic.has_key(key): #判斷這個key是否在原有的字典中
old=dic[key]
del(dic[key])
dic[key]=int(old)+int(new)
#print dic
for tran in dic:
out.write(tran+'\t'+str(dic[tran])+'\n')
out.flush()
out.close()
5.后續找出每個基因的所有轉錄本,用heapq庫找出最長的一個。庫用法https://blog.csdn.net/Cassiel60/article/details/88344137
同樣是解析這個文件,可以看出文件中的id是根據第三列進行編號的,沒有實際意義,只是可以看出共有多少個gene、exon、cds等。不過在第三列為gene時,Name=和Dbxref=GeneID: 與第四列為exon時,Dbxref=GeneID:和transcript_id=進行基因與轉錄本的正確匹配。可以在上面代碼中的字典加入Dbxref=,字典中一鍵對應多個值。
6.得到的兩個文件進行merge,就可以得到基因,轉錄本,長度的文件了。
?
更多文章、技術交流、商務合作、聯系博主
微信掃碼或搜索:z360901061

微信掃一掃加我為好友
QQ號聯系: 360901061
您的支持是博主寫作最大的動力,如果您喜歡我的文章,感覺我的文章對您有幫助,請用微信掃描下面二維碼支持博主2元、5元、10元、20元等您想捐的金額吧,狠狠點擊下面給點支持吧,站長非常感激您!手機微信長按不能支付解決辦法:請將微信支付二維碼保存到相冊,切換到微信,然后點擊微信右上角掃一掃功能,選擇支付二維碼完成支付。
【本文對您有幫助就好】元
